This study, published in Nature Communications and led by the Zhong Qing team at Shanghai Jiao Tong University School of Medicine, for the first time fully elucidates the molecular mechanism of Necrosis by Sodium Overload (NECSO): Na⁺ is not a passive consequence of cell death, but triggers cell death by disrupting mitochondrial energy metabolism. Na⁺ acts as an active initiator of necrosis, rather than merely a late participant in osmotic disruption.

Research Object: Necrosis by Sodium Overload (NECSO).
Core Initiation: Small molecule NC1 persistently activates TRPM4 channel → massive Na⁺ influx.
Core Hub: Mitochondrial NCLX (Na⁺/Ca²⁺ exchanger).
Core Outcome: Mitochondrial energy collapse → Na/K ATPase inactivation → ion gradient collapse → cell osmotic swelling and lysis.
Scientific Breakthrough: Na⁺ is an active lethal signal rather than just an osmotic effect; the TRPM4-NCLX-mitochondrial energy axis is defined as the complete pathway for NECSO.
| Traditional View | New Discovery (This Study) |
|---|---|
| Na⁺ influx is a late result of cell death (after membrane rupture) | Na⁺ influx is an active initiator of cell death |
| Na⁺ causes cell death mainly via osmotic imbalance | Na⁺ actively induces necrosis by inhibiting mitochondrial energy production |
| Na⁺ is a "passive executor" | Na⁺ is an "active regulator" |
(1) NC1 activates TRPM4 (monovalent cation channel), inducing Na⁺ overload necrosis, named NECSO.
(2) NECSO is independent of apoptosis, necroptosis, ferroptosis, and pyroptosis; unaffected by classical pathway inhibitors/knockout, and completely blocked by TRPM4 knockout.
(1)How does Na⁺ influx lead to cell death? What role do mitochondria play in NECSO?
(2)How are ionic homeostasis and energy metabolism coupled?
(3)What is the execution mechanism of final membrane rupture?
(1) Temporal evidence: Na⁺ influx → early energy depletion → late membrane rupture (causal establishment)
NC1 treatment for 30 minutes: significant increase in intracellular Na⁺;
2–4 hours: ATP levels drop sharply, with PI positivity < 10%;
18 hours: PI is nearly 100%, LDH release occurs later → energy depletion precedes membrane damage, which is the cause of death rather than the effect
(2) Na⁺ dependency verification
In the absence of Na⁺ medium (NMDG⁺ substitution): NC1 did not increase intracellular Na⁺ levels, ATP levels did not decrease, and there was no cell death;
Replenishment of Na⁺: Recovery from death → Na⁺ influx is a necessary condition for energy collapse and NECSO;
Salvage experiment: Creatine, α-lipoic acid (promotes ATP production) → inhibits NC1-induced LDH release → energy supplementation can block NECSO.
Proof: Energy depletion is the early core event of NECSO, rather than a secondary result following membrane rupture.
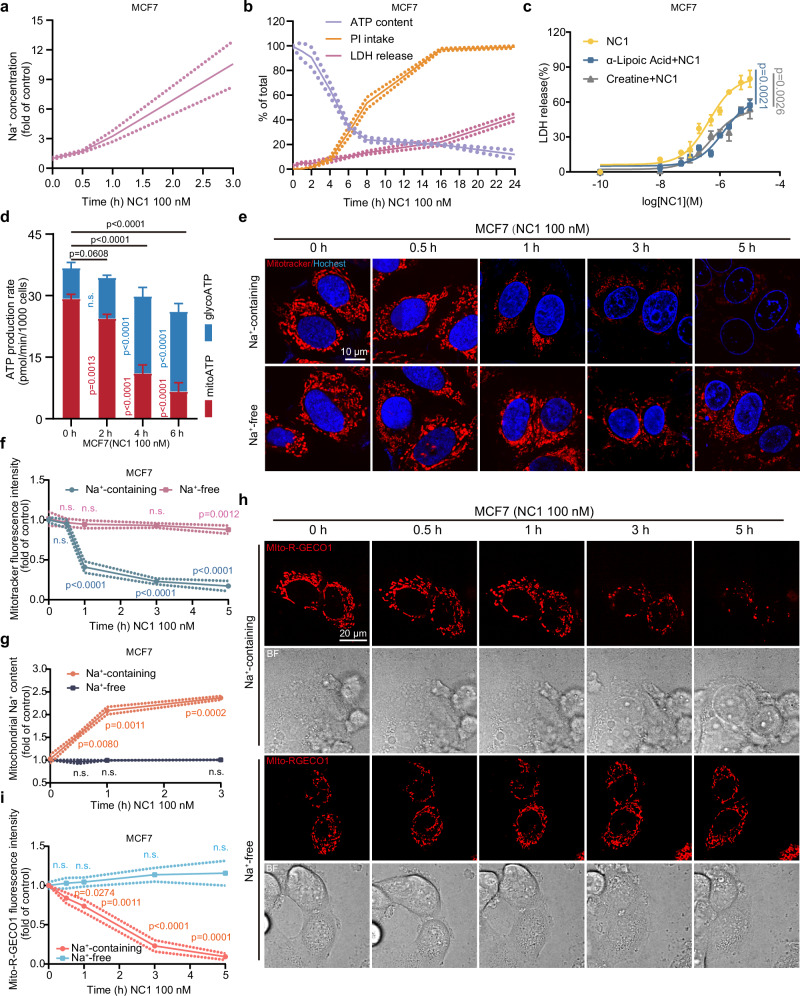
Figure 1. NC1-induced energy depletion and mitochondrial ion imbalance are dependent on sodium ion influx.
(1) Mitochondrial structure: Rapid swelling and cristae rupture after NC1 treatment; absent in Na⁺-free conditions.
(2) Mitochondrial ionic imbalance:
Cytosolic Na⁺ ↑ → enters mitochondria via NCLX → mitochondrial Na⁺ ↑;
Simultaneous NCLX exchange → mitochondrial Ca²⁺ ↓.
(3) Metabolism & respiration
Oxidative phosphorylation (OXPHOS) inhibited, mitochondrial ATP (mitoATP) sharply reduced;
Glycolysis increased compensatorily, but total ATP still decreased;
High Na⁺ directly inhibits respiratory chain complex II+III activity.
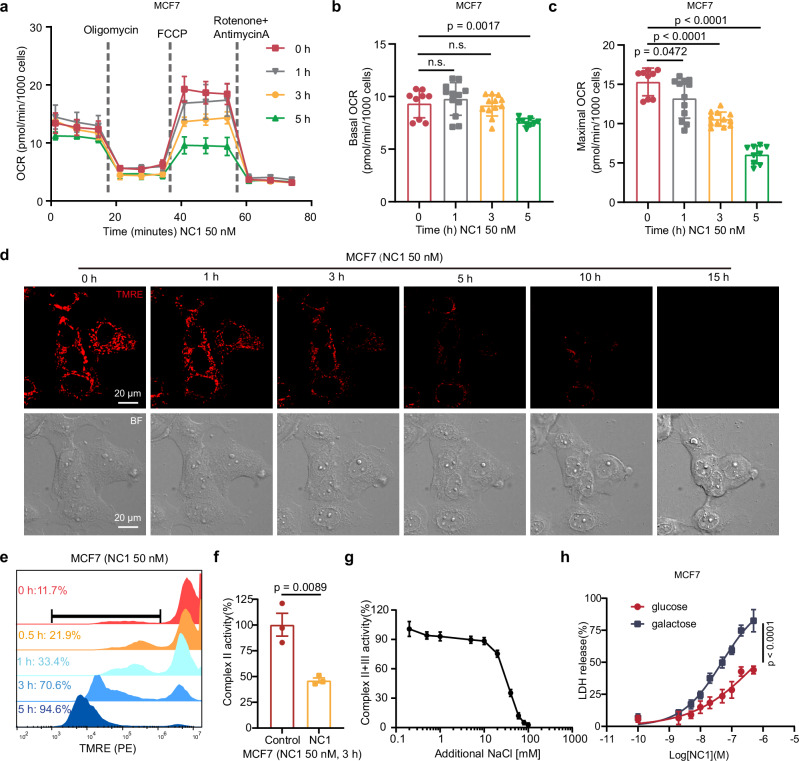
Figure 2. Mitochondrial respiration is inhibited in NECSO.
(1) Key Findings:
Accumulation of intermediate products in the TCA cycle: isocitrate, α-ketoglutarate, succinate, fumarate, malate;
Time dependence: Accumulates as the treatment time with NC1 increases.
(2) The ratio of succinate to fumarate progressively increases (from 0 to 3 hours: approximately 3-fold to approximately 12-fold), confirming the inhibition of succinate dehydrogenase (SDH/Complex II) function.
(3) Pyruvate metabolism disorder:
Pyruvate accumulation;
Accumulation of thiamine pyrophosphate (TPP, a cofactor of PDHc);
Increased phosphorylation of PDHA1 (E1α subunit) at Ser293 → Inactivation of PDHc.
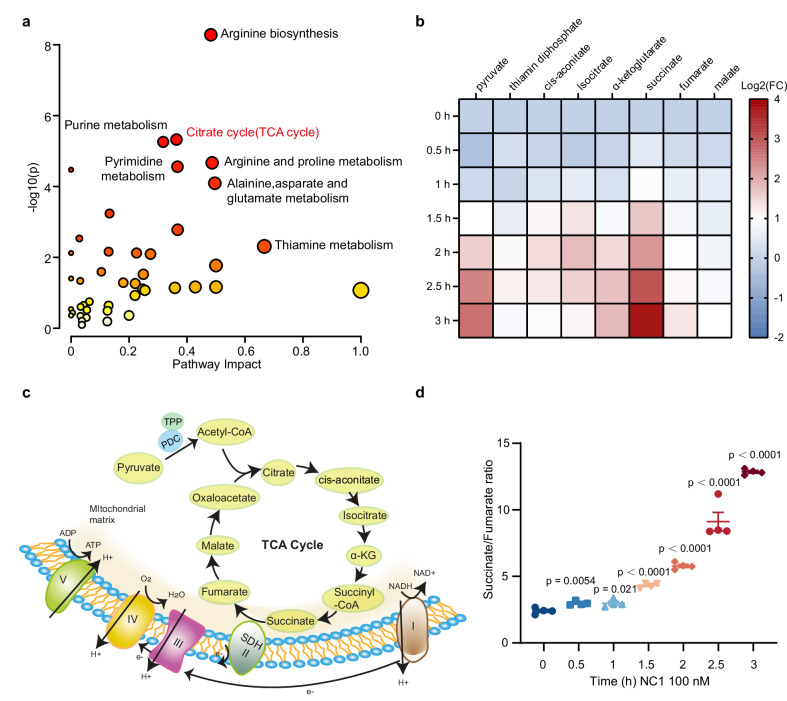
Figure 3. Inhibition of TCA cycle in NECSO.
| Detection index | NC1 alone | NC1 + CGP | Effect |
|---|---|---|---|
| Mitochondrial swelling (TEM) | Severe | Significantly alleviated | Protection |
| Mitochondrial Na⁺ | ↑ ↑ | Reversed | Blocked |
| Mitochondrial Ca²⁺ | ↓ ↓ | Restored | Reversed |
| Membrane potential (TMRE) | Collapsed | Maintained | Protection |
| Maximal respiration | ↓ ↓ | Restored | Reversed |
| Complex II+III activity | ↓ ↓ | Partially restored | Improved |
| ATP production rate | ↓ ↓ | Restored | Reversed |
| Cell death (LDH) | ++++ | + | Significantly inhibited |
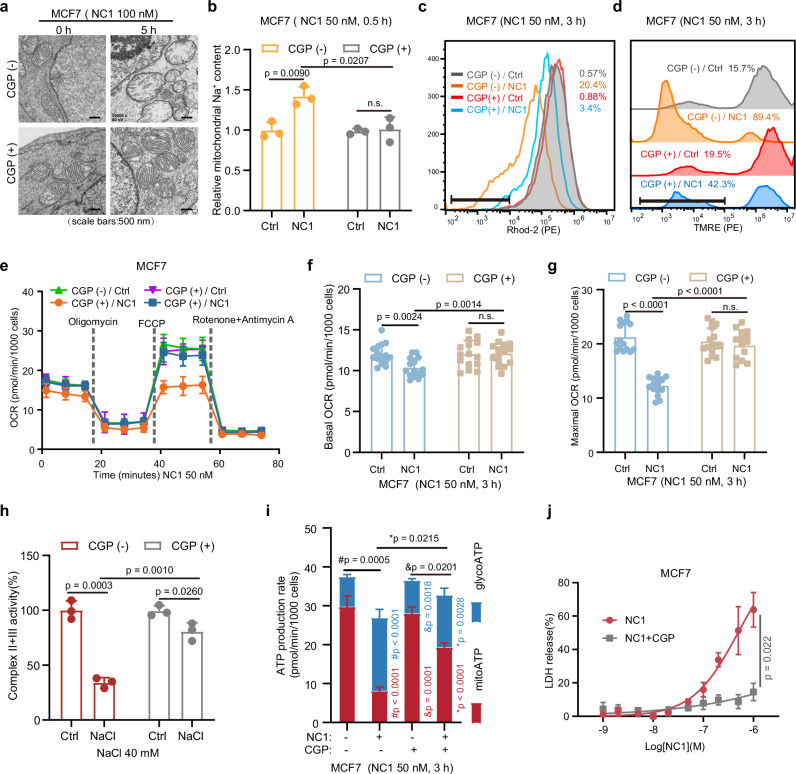
Figure 4. Inhibiting NCLX reverses NC1-induced mitochondrial dysfunction.
(1) Na/K-ATPase activity: Began to decrease 1 h after NC1 treatment, further inhibited over time.
(2) Ouabain (Na/K-ATPase inhibitor) effect:
Exacerbate NC1-induced Na⁺ accumulation and K⁺ loss;
Even at an extremely low concentration (10 nM), it can significantly promote NC1-induced cell death;
Demonstrate that Na/K-ATPase inhibition is a crucial step in the execution of NECSO.
(3) Verification of Na⁺-free conditions:
In the absence of extracellular Na⁺, NC1 does not inhibit Na/K-ATPase activity;
It has been confirmed that Na⁺ influx is a prerequisite for Na/K-ATPase inhibition.
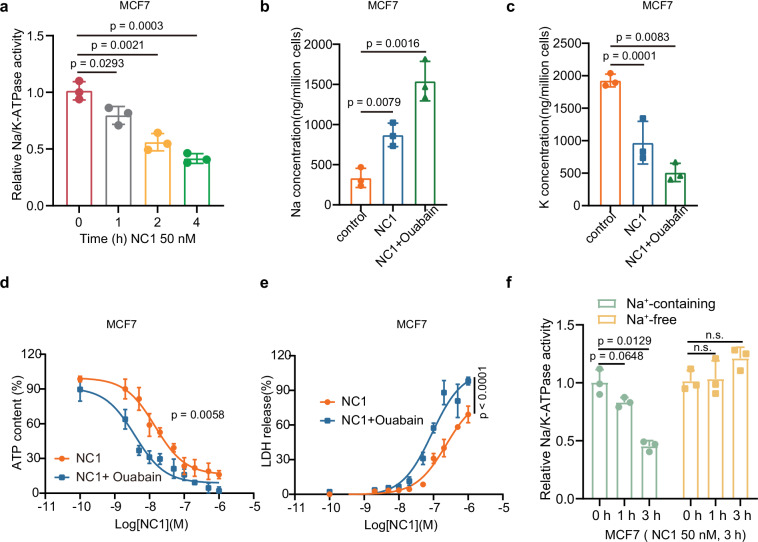
| Detection index | Wild-type (WT) | TRPM4-KO |
|---|---|---|
| Cytosolic Na⁺ (CoroNa Green) | Significantly ↑ after NC1 | No change |
| Mitochondrial Na⁺ | ↓ after NC1 | No change |
| Mitochondrial Ca²⁺ | ↓ after NC1 | No change |
| Mitochondrial morphology (TEM) | Swollen | Normal |
| Membrane potential | Collapsed | Normal |
| OCR (basal/maximal) | ↓ ↓ | Normal |
| Complex II activity | ↓ | Normal |
| ATP production rate | ↓ ↓ | Normal |
| Na/K-ATPase activity | ↓ | Normal |
| Cell death (LDH) | ++++ | Almost none |
Key conclusions:
NC1 does not act directly on mitochondria;
All effects are dependent on TRPM4-mediated Na⁺ influx;
Establish a complete signal chain: NC1 → TRPM4 → Na⁺ influx → NCLX → mitochondrial dysfunction.
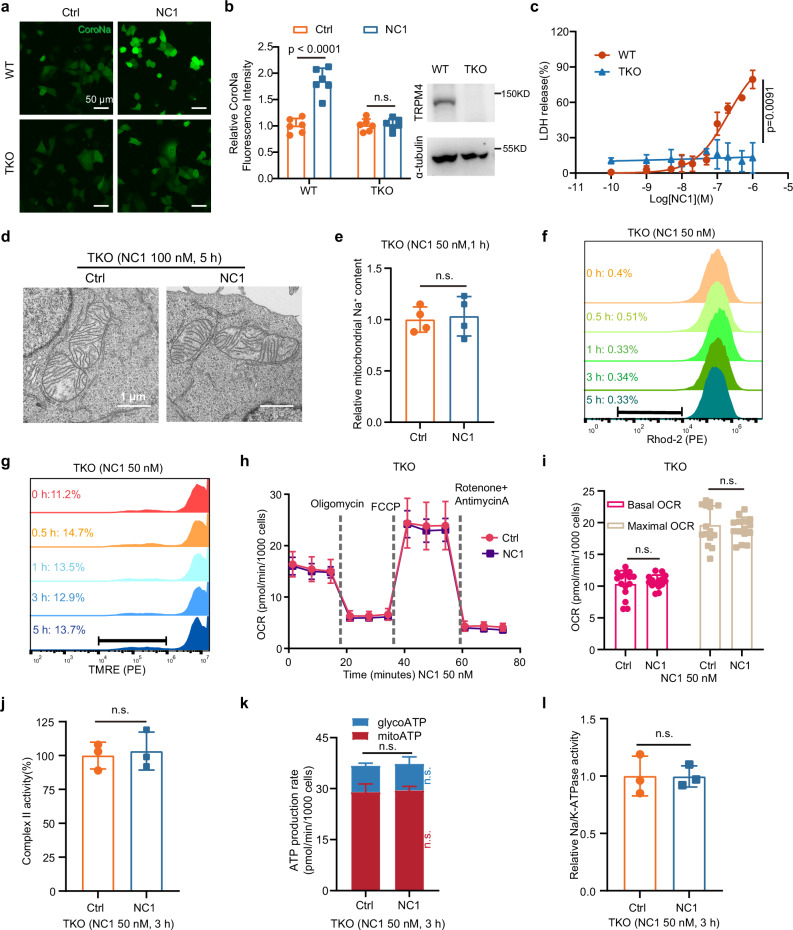
Figure 6. TRPM4 is essential for NC1-induced mitochondrial dysfunction.
(1) The experiment on osmoprotectants reveals the principle of membrane pore formation:
Ion gradient collapse → increased osmotic pressure → water influx → cell swelling;
Formation of nanoscale defects in the membrane → leakage of macromolecules.
Experimental results: PEG 8K almost completely blocked NC1-induced LDH release, cell swelling, and PI uptake, demonstrating the presence of variable-sized membrane defects in NECSO.
| Osmoprotectant | Hydration Diameter | LDH Release Inhibition |
|---|---|---|
| Sucrose | ~0.9 nm | Partial |
| PEG 2K | ~2.6 nm | Good |
| PEG 4K | ~3.2 nm | Better |
| PEG 8K | ~4 nm | Almost complete inhibition |
(2) AQP4 involvement in water influx AQP4 inhibition experiment:
AER-271 (AQP4 small molecule inhibitor): significantly reduced LDH release;
AQP4 siRNA: both independent sequences significantly reduced cell death;
Suggesting that AQP4-mediated water influx is involved in the execution of NECSO.
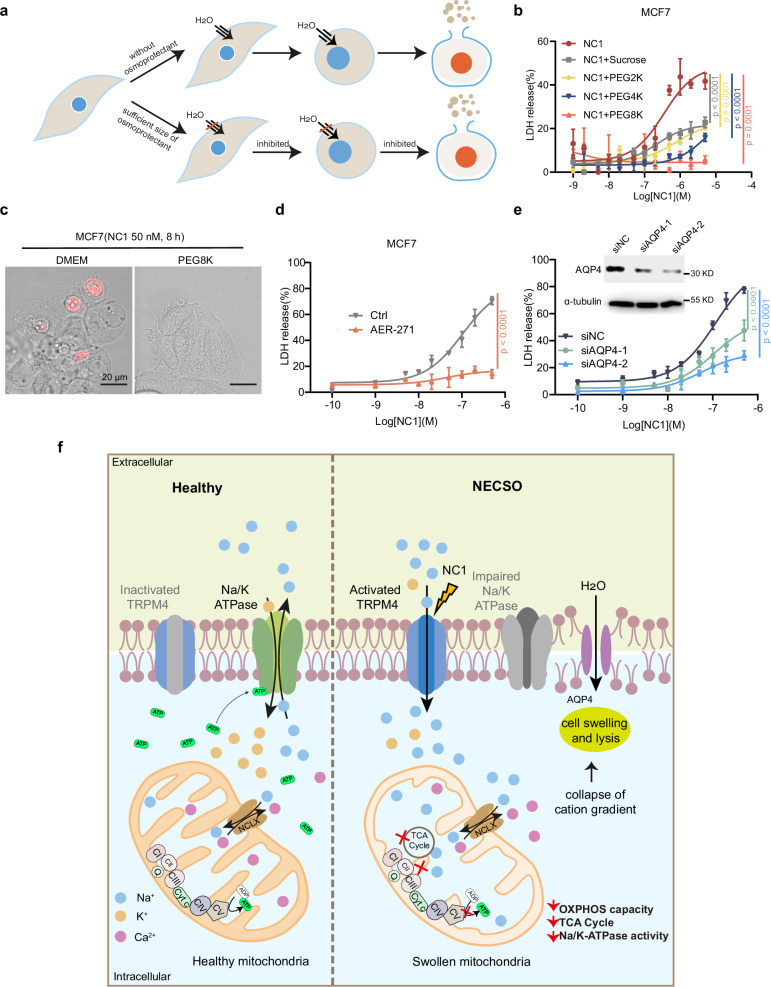
Figure 7. Osmoprotectants and AQP4 inhibitors can prevent membrane rupture in NECSO.
The study indicates that sodium influx can regulate cell death by inhibiting mitochondrial energy production. Mechanistically, the study reveals that TRPM4-dependent NC1-induced sodium influx can activate NCLX-mediated transport, leading to an increase in [Na+] and a decrease in [Ca2+]. This perturbation hinders the tricarboxylic acid cycle, mitochondrial oxidative phosphorylation, and overall energy production. Consequently, the activity of the sodium-potassium pump is impaired, ultimately resulting in the collapse of intracellular cation gradients, promoting osmotic cell swelling and membrane rupture.
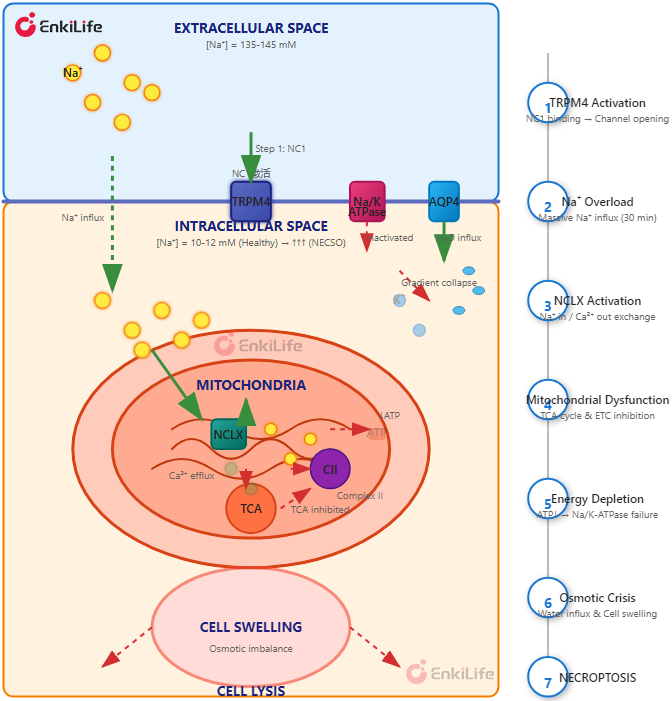
Sodium disrupts mitochondrial energy metabolism to execute NECSO. Qiao Y, et al. Nat Commun. 2025. [PMID: 41390760]
Related Products
