Literature Sharing: Microglia-specific Regulation of Lipid Metabolism in Alzheimer's Disease
I. Research Background
Alzheimer's disease (AD), as the most common neurodegenerative disease and major cause of dementia, is characterized by severe disruption of brain homeostasis as one of its core pathological features. The role of abnormal lipid metabolism in disease progression has long been insufficiently recognized. In recent years, with the development of lipidomics technology, studies have found that imbalances in lipids such as phospholipids, sphingolipids, and cholesterol may be initiating factors in AD pathogenesis, rather than mere pathological byproducts. These lipid disorders can promote amyloidogenesis through mechanisms such as regulating membrane fluidity, secretase compartmentalization, and Aβ aggregation kinetics. The study "Microglia-specific regulation of lipid metabolism in Alzheimer's disease revealed by microglial depletion in 5xFAD Mice" focuses on microglia—the brain's resident immune cells. Combining the 5xFAD transgenic mouse model with human post-mortem brain tissue samples, the study investigates the specific regulatory role of microglia in lipid metabolism disorders in AD. The aim is to decipher the association between abnormal lipid metabolism in AD and different brain cell types and molecular pathways, providing new therapeutic targets for the disease. Previous studies have confirmed that lipid metabolism risk genes enriched in microglia (such as TREM2, PLCG2, GRN, etc.) are involved in Aβ clearance and neuroinflammation regulation. However, the specific impact of microglial depletion on brain lipid metabolism has been widely overlooked, with only one study focusing on its necessity for leukotriene synthesis, which became the core entry point for this study.
II. Research Methods
The study uses the 5xFAD transgenic mouse model, which recapitulates key features of AD amyloid pathology, covering both B6SJL and B6 genetic backgrounds. Through both pharmacological and genetic microglial depletion approaches, the study distinguishes between microglia-dependent and microglia-independent lipid metabolism disorder mechanisms in AD. Pharmacological depletion uses the CSF1R inhibitor PLX5622, administered to 6-week-old 5xFAD and normal control (Non-Tg) mice for short-term (2 weeks) and long-term (3.5 months) feeding. Genetic depletion involves crossing Csf1r enhancer deletion mice with 5xFAD mice to obtain four genotypes, which are harvested at 5 months of age. The study combines shotgun lipidomics, RNA profiling (NanoString glial profiling panel), and immunofluorescence techniques to detect changes in lipid species, gene expression, and pathological markers in mouse brains. Additionally, post-mortem brain tissues from 10 non-disease controls and 10 AD patients are selected for lipidomic analysis, adjusting for confounding factors such as sex and gray-white matter ratio to validate the clinical relevance of mouse model results. Finally, statistical analyses (PLS-DA, volcano plots, etc.) are used to identify significant lipid changes and their correlations with pathological features.
III. Results Analysis
1. Validation of Microglial Depletion Strategy and Analysis of Amyloid Pathology Phenotype
To clarify the role of microglia in AD lipid metabolism, the study first validated the effectiveness of both depletion methods and observed their effects on amyloid pathology. Short-term PLX5622 treatment almost completely eliminated microglia in both 5xFAD and Non-Tg mice, reducing P2Y12 immunoreactive areas by 99%, while long-term treatment reduced microglia by 82% in 5xFAD mice. The remaining microglia were mostly disease-associated microglia (DAM) surrounding amyloid plaques, which showed significantly higher P2Y12 immunofluorescence intensity than non-plaque microglia and exhibited a more amoeboid morphology with fewer processes. At 2 months of age, 5xFAD mice had almost no amyloid deposits in the brain, while at 5 months, Aβ and fibrillar plaque load increased significantly. Long-term PLX5622 treatment slightly reduced Aβ load and fibrillar plaque load, consistent with previous studies. Notably, amyloidosis significantly increased astrocyte reactivity, as indicated by elevated GFAP immunoreactive area and brightness. However, PLX5622 treatment did not affect GFAP intensity or immunoreactive area (after correcting for Aβ load), nor did it alter brain APP protein levels or synaptic density. In the genetic depletion model, microglia were absent from the brains of Csf1r-deficient mice, and 5xFAD mice with genetic depletion showed similar amyloid pathology changes to those with pharmacological depletion.
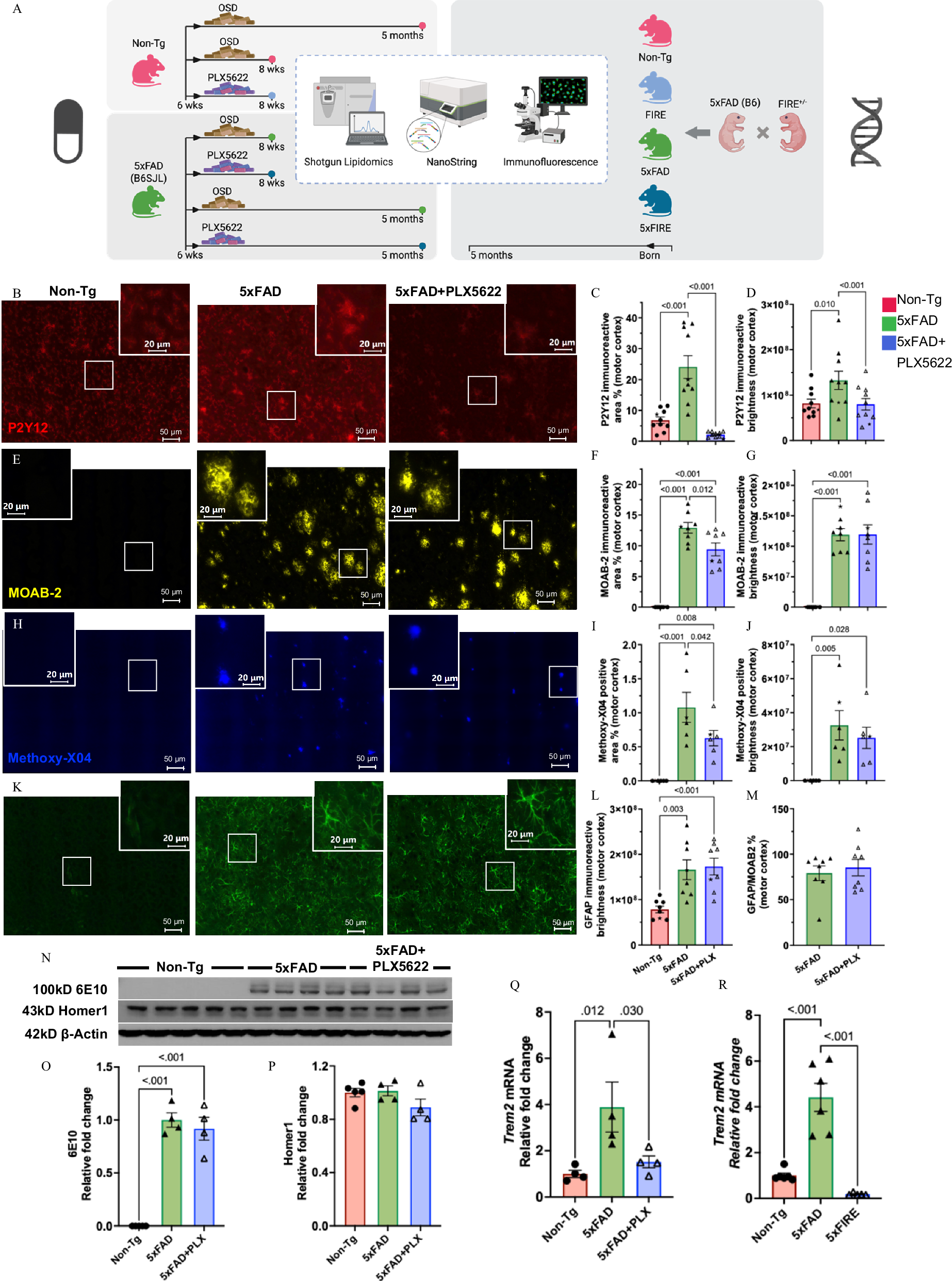
2. Differential Effects of Amyloidosis and Microglial Depletion on Brain Lipidome and Transcriptome
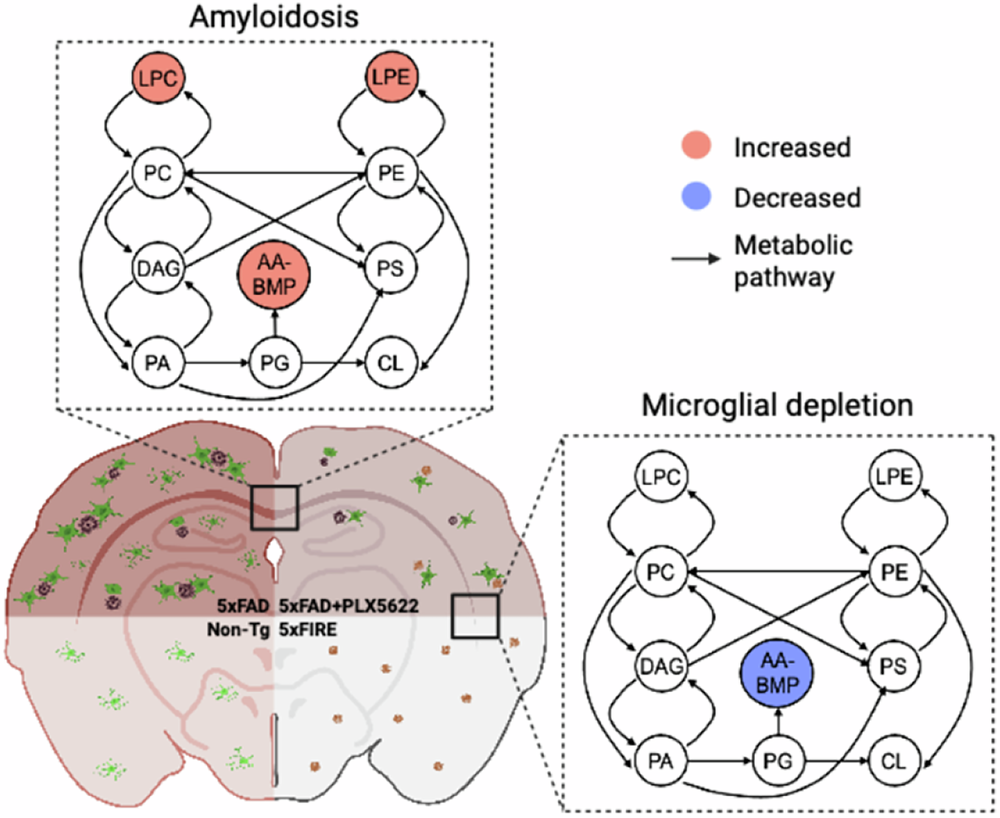
Through analysis of 198 lipid species, the study found that amyloidosis causes significant characteristic changes in the brain lipidome. Both in the pharmacological and genetic depletion groups, the lipidomes of Non-Tg and 5xFAD mice could be clearly distinguished by PLS-DA analysis. Consistent increases in LPC, LPE, and BMP species were observed in both cohorts, suggesting these lipids are closely associated with AD amyloid pathology. More importantly, microglial depletion caused differential changes in the lipidome: long-term pharmacological depletion led to a more significant shift in the lipidome, showing complete PLS-DA separation from the 5xFAD group, and significantly increased multiple lysophospholipid species; while genetic depletion uniquely reduced myelin lipids and lipids related to energy metabolism, suggesting different depletion degrees have different effects on brain lipid metabolism. However, both depletion methods shared a key finding—both prevented the accumulation of bis(monoacylglycerol) phosphate containing arachidonic acid (20:4) (AA-BMP), suggesting that AA-BMP accumulation is highly dependent on microglia. Transcriptomic analysis further supported this conclusion: the transcriptomes of each group in both cohorts could be clearly distinguished by PLS-DA. The transcriptome of 5xFAD mice with complete microglial depletion showed greater differences from Non-Tg mice than 5xFAD mice without depletion, suggesting that complete microglial depletion induces more significant transcriptomic remodeling. Amyloidosis-induced inflammation-related genes were downregulated by microglial depletion in both cohorts, with more significant downregulation in the genetic depletion group. Venn analysis revealed that genes commonly downregulated by both depletion methods were primarily involved in immune response and lysosomal function pathways.
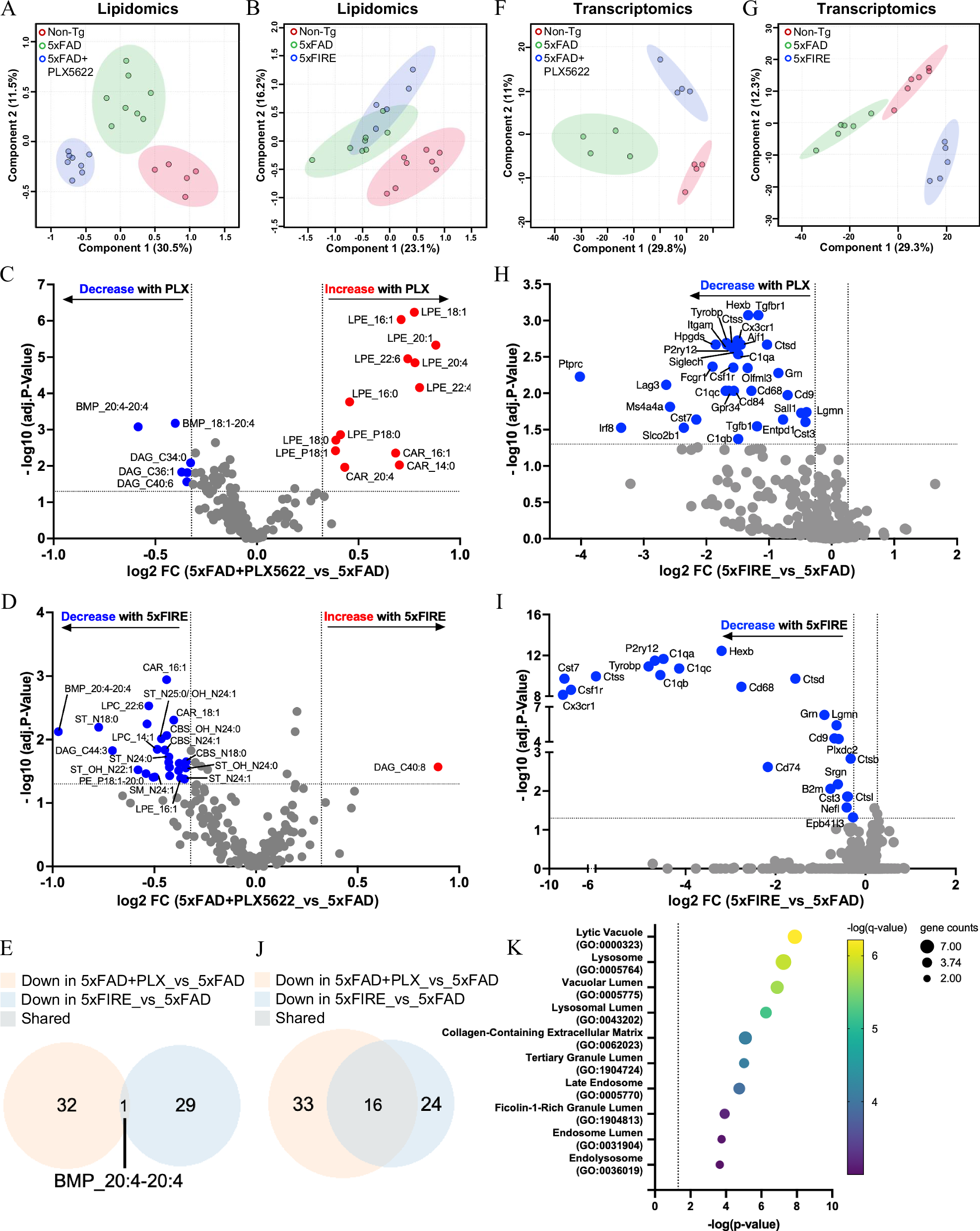
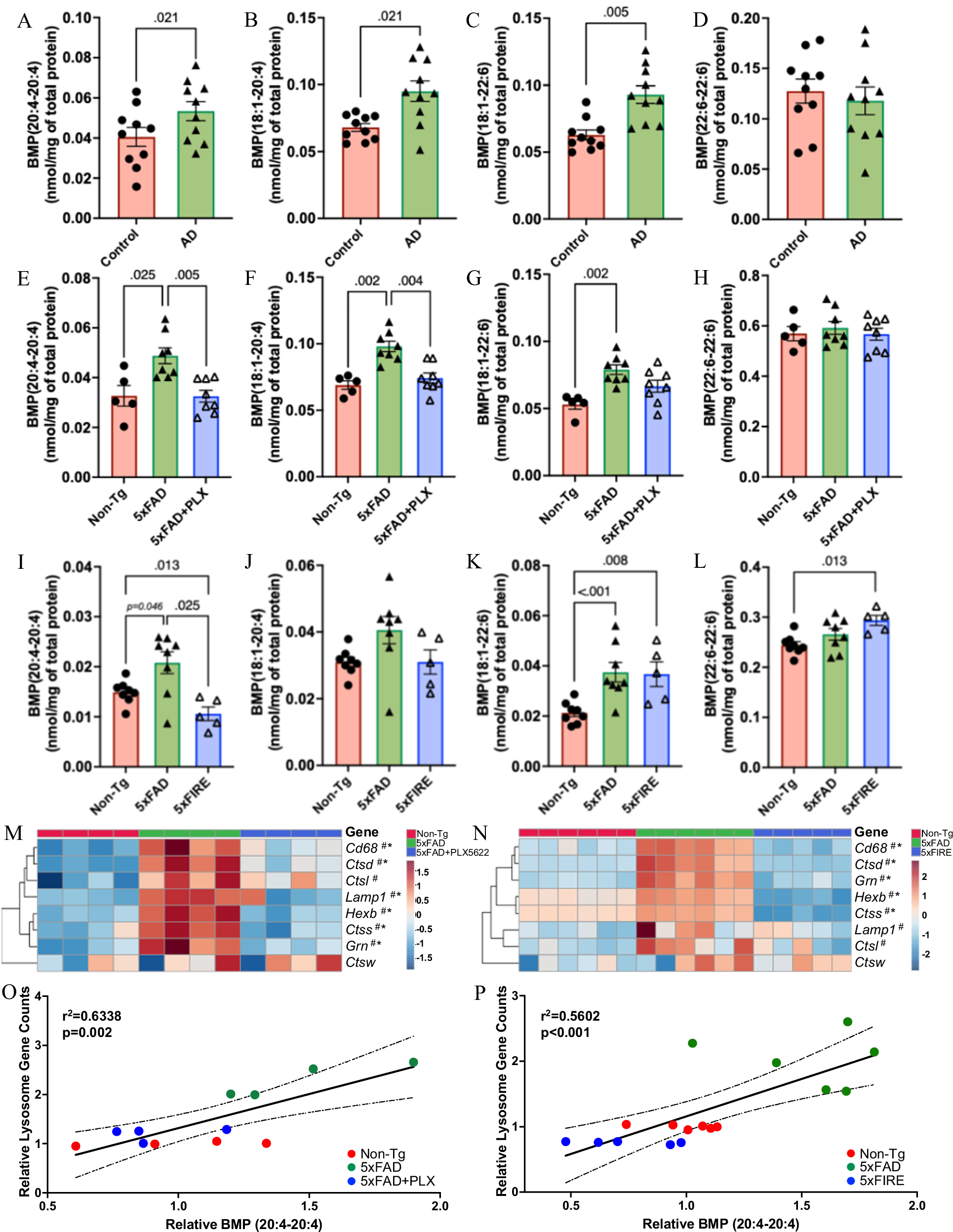
3. Microglial Depletion Prevents Amyloidosis-induced Lysosomal AA-BMP Accumulation
Given that AA-BMP was the only lipid species affected by both depletion methods, the study further investigated its changes in AD and its association with microglia. In human AD post-mortem brain tissue, abnormal BMP accumulation was detected, highly consistent with the trend in the 5xFAD mouse model—BMP species containing arachidonic acid (20:4) and oleic acid (18:1) were significantly increased, while BMP species containing docosahexaenoic acid (DHA, 22:6) showed no significant changes. This result confirmed the clinical relevance of abnormal BMP accumulation in AD and demonstrated that the mouse model can well simulate the lipid pathological features of human AD. In the mouse model, although microglial intervention did not significantly change overall BMP levels, different types of BMP molecules showed distinct response characteristics to microglial depletion. Among them, arachidonic acid-containing BMP subtypes were highly sensitive to microglial depletion, showing significant decreases after long-term pharmacological intervention; in the genetic microglial knockout model, these two BMP subtypes also showed significant reduction or similar downward trends. In sharp contrast, DHA-containing BMP subtypes only accumulated with amyloid pathology progression, and their levels did not decrease due to microglial depletion. The subtype completely composed of DHA even showed an increasing trend, suggesting that different BMP subtypes are regulated by different mechanisms.
Further analysis found that amyloidosis induces abnormal upregulation of lysosome-related genes, and both microglial depletion methods could restore the expression of these genes to levels close to those in normal mice. The protein level of CD68, a microglia/macrophage-specific lysosomal marker, was also significantly increased in 5xFAD mouse brains, but decreased significantly after long-term pharmacological microglial depletion. As another important indicator of lysosomal function, abnormally glycosylated LAMP1 induced by amyloid pathology was significantly improved after microglial depletion, while normal molecular weight LAMP1 was not affected, indicating that microglial depletion can partially alleviate lysosomal dysfunction in AD. BMP, as a characteristic lipid localized in the late endosome-lysosome system, its levels can directly reflect lysosomal function status. Further analysis showed that arachidonic acid-containing BMP was highly positively correlated with the expression levels of lysosome-related genes, confirming at the molecular level the core mechanism by which microglia regulate specific BMP metabolism through lysosomal function.
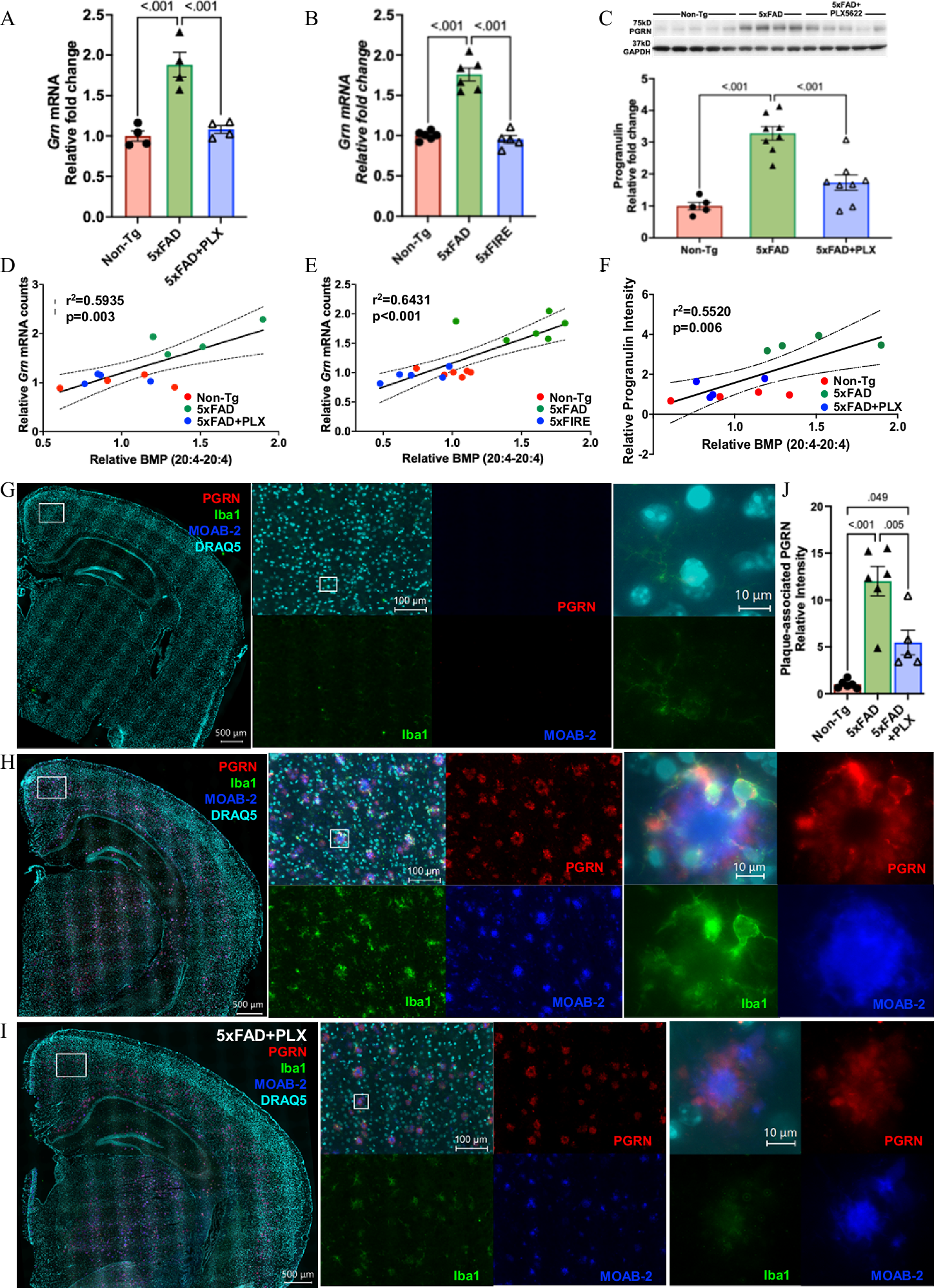
4. PGRN May Be a Potential Regulator of AA-BMP
Based on transcriptomic analysis and existing research, the study focused on progranulin (PGRN), a protein closely related to lysosomal function. This protein is associated with neurodegenerative diseases such as frontotemporal dementia and amyotrophic lateral sclerosis, and clinical studies have confirmed its association with AD. Experiments found that GRN expression levels were significantly higher in 5xFAD model mice than in normal control mice, and both long-term pharmacological and genetic microglial depletion treatments effectively inhibited the amyloidosis-induced upregulation of GRN expression. At the protein level, long-term pharmacological depletion of microglia also significantly alleviated the abnormal accumulation of PGRN protein induced by amyloid pathology.
Immunofluorescence results showed that PGRN is mainly expressed in microglia and neurons in the brain, with the highest expression in reactive DAM surrounding amyloid plaques. It colocalizes with microglial markers such as Iba1 and CD68. Even after PLX5622 treatment, the remaining plaque-associated DAM still showed high PGRN expression. Microglial depletion significantly reduced plaque-associated PGRN accumulation, which was closely related to the reduction in plaque load and surrounding microglia. Correlation analysis further confirmed that PGRN mRNA and protein levels were highly correlated with AA-BMP levels. Combined with previous studies showing BMP deficiency in GRN knockout mouse brains, it was clear that microglia-derived PGRN is a potential regulator of AA-BMP metabolism, revealing the specific molecular mechanism by which microglia regulate AA-BMP and closely linking the GRN gene to lipid metabolism disorders in AD.
5. Microglia Do Not Drive Amyloidosis-induced Lysophospholipid Accumulation
In addition to BMP, lysophosphatidylcholine (LPC) and lysophosphatidylethanolamine (LPE) are potential biomarkers for AD, but their accumulation mechanisms remain unclear. LPC is mainly produced by phospholipase A2 hydrolysis of phosphatidylcholine, a core component of cell membranes, and different subtypes of LPC have completely different functions. Saturated and monounsaturated LPC have typical pro-inflammatory effects, promoting chemokine release and reactive oxygen species generation, exacerbating neuroinflammation; while polyunsaturated LPC has anti-inflammatory effects, counteracting the damage caused by pro-inflammatory subtypes. This study confirmed that total LPC levels are significantly increased in both human AD brain tissue and amyloidosis mouse models, driven by the upregulation of multiple subtypes. More importantly, in the amyloidosis environment, neither long-term pharmacological treatment nor genetic knockout to deplete microglia could change total LPC content, only genetic knockout slightly reduced polyunsaturated anti-inflammatory LPC, suggesting weakened anti-inflammatory regulation ability. This result clearly indicates that amyloidosis-induced pro-inflammatory LPC accumulation is completely independent of microglia.
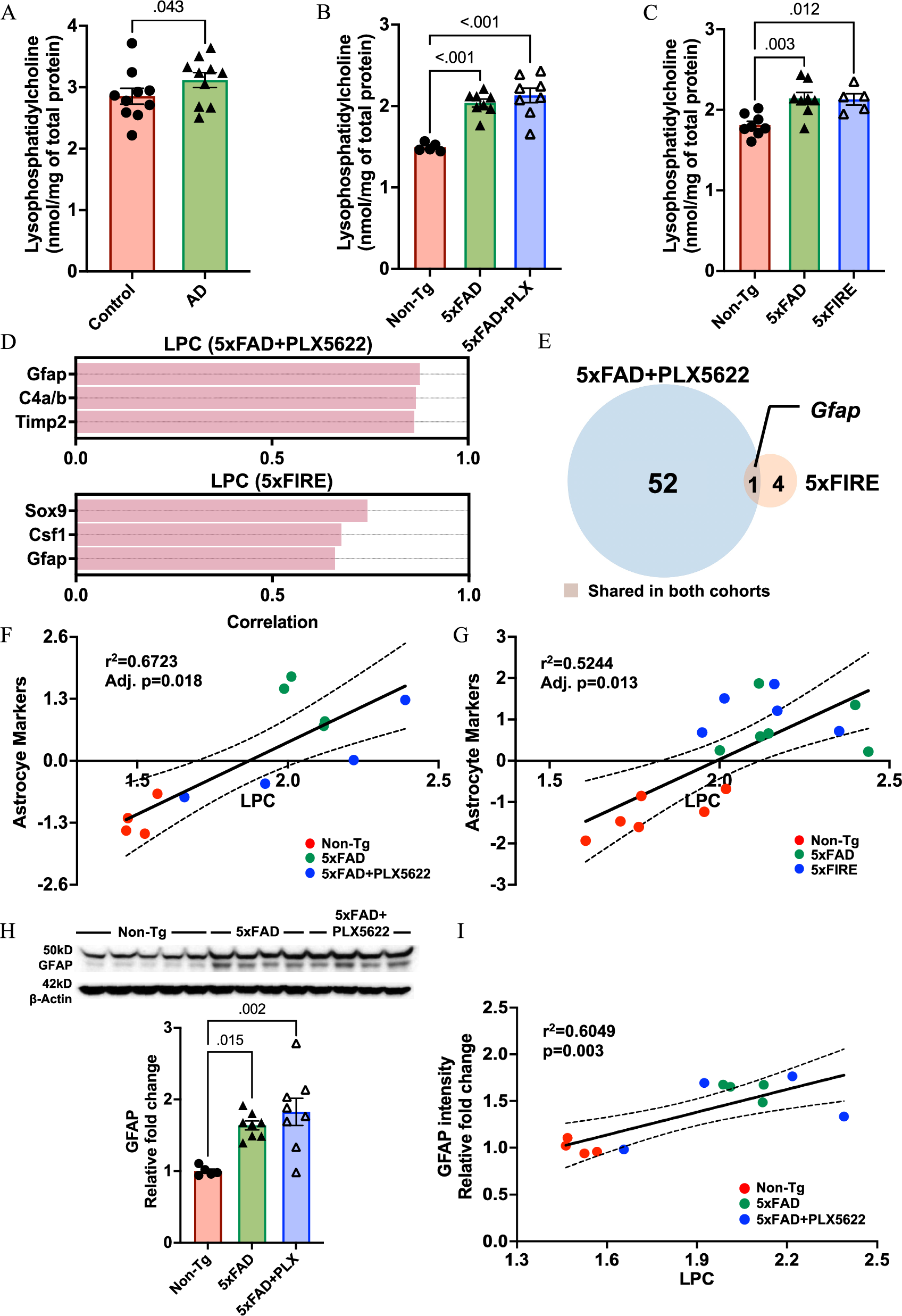
As another key lysophospholipid, LPE is produced by hydrolysis of phosphatidylethanolamine, an important component of cell membranes and organelle membranes. It is involved in calcium signal regulation, has anti-apoptotic activity, and promotes neuronal differentiation and migration, but its abnormal accumulation is highly correlated with oxidative stress. The study found that LPE was also consistently increased in AD patient brain tissue and amyloidosis mouse models, showing obvious subtype-specific changes. Surprisingly, in the amyloidosis background, long-term partial depletion of microglia using PLX5622 further exacerbated LPE accumulation, with the most prominent increase in non-plasmalogen LPE, which can be degraded from plasmalogen under reactive oxygen species attack. Although no clear evidence of oxidative stress driving LPE accumulation was directly detected in this study, it was found that after microglial depletion, the expression of the core antioxidant regulator Nrf2 decreased and was negatively correlated with LPE levels, suggesting that microglial depletion weakens the body's antioxidant response, thereby indirectly promoting abnormal LPE increase. Unlike pharmacological treatment, genetic microglial depletion did not further increase LPE. Meanwhile, in the absence of amyloidosis, complete microglial depletion also caused abnormal accumulation of LPC and LPE, while short-term microglial depletion had no significant effect, which is related to the milder early pathological stage.
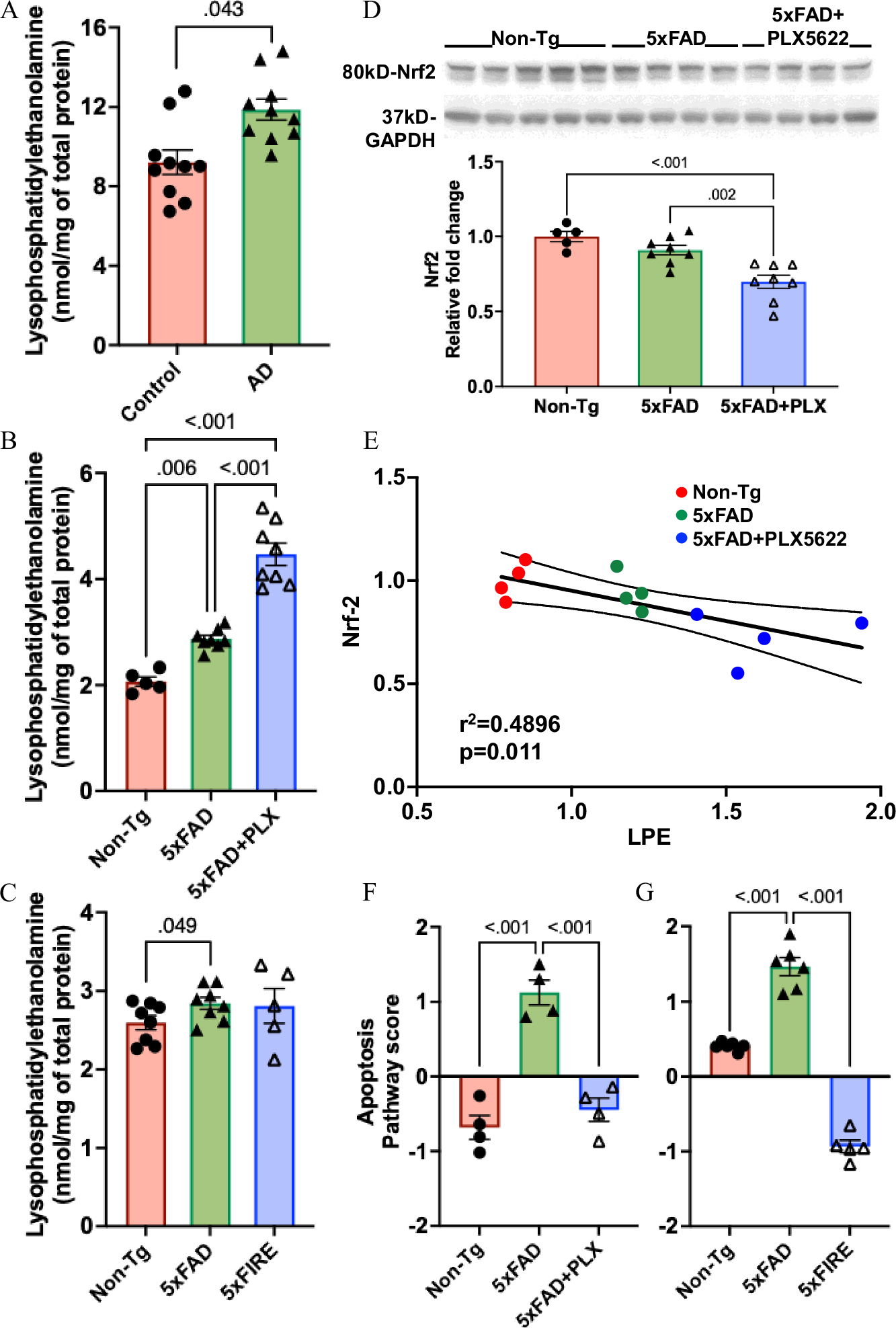
Based on the above results, it can be concluded that during Alzheimer's disease progression, the abnormal accumulation of LPC and LPE is not primarily driven by microglia. LPC is mainly associated with astrocyte activation, while LPE is more regulated by oxidative stress. This finding provides important insights into understanding the cell-specific mechanisms of lipid disorders in AD.
IV. Conclusion
Through rigorous experimental design, combining mouse models with human samples, this study systematically revealed for the first time the specific regulatory role of microglia in AD lipid metabolism, clarifying the regulatory mechanisms and cellular sources of different lipid species. Microglia regulate lysosomal function through PGRN, thereby specifically controlling AA-BMP accumulation, and this regulation is independent of amyloidosis; while the accumulation of LPC and LPE depends on astrocyte activation and oxidative stress, independent of microglia. The study not only linked lipid metabolism disorders in AD to specific brain cell types and molecular pathways, solving the long-standing confusion about lipid metabolism regulatory mechanisms in AD, but also connected the lipid accumulation initially observed by Alois Alzheimer with gene-defined lipid pathways, providing a new target for AD treatment—microglial lipid homeostasis, especially the regulation of AA-BMP and PGRN, which is expected to become a potential strategy for improving AD pathological progression. At the same time, this study also provides new ideas for subsequent AD lipid metabolism research, that is, analyzing the mechanisms of lipid disorders from a cell-specific perspective, laying the foundation for precise treatment.
References
Xu Z, Kiani Shabestari S, Barannikov S, Bieniek KF, Blurton-Jones M, Palavicini JP, Han X. Microglia-specific regulation of lipid metabolism in Alzheimer's disease revealed by microglial depletion in 5xFAD Mice. Nat Commun. 2025 Oct 15;16(1):9156. doi: 10.1038/s41467-025-64161-z. PMID: 41093842; PMCID: PMC12528747.
EnkiLife Products:
Western Blot Complete Experimental Protocol
TSA Multiplex Fluorescence Kit