Literature Sharing: TKI Combined with CTLA4 Blockade: A Novel Mechanism Reshaping the Immune Microenvironment of Lung Adenocarcinoma Brain Metastasis
I. Research Background
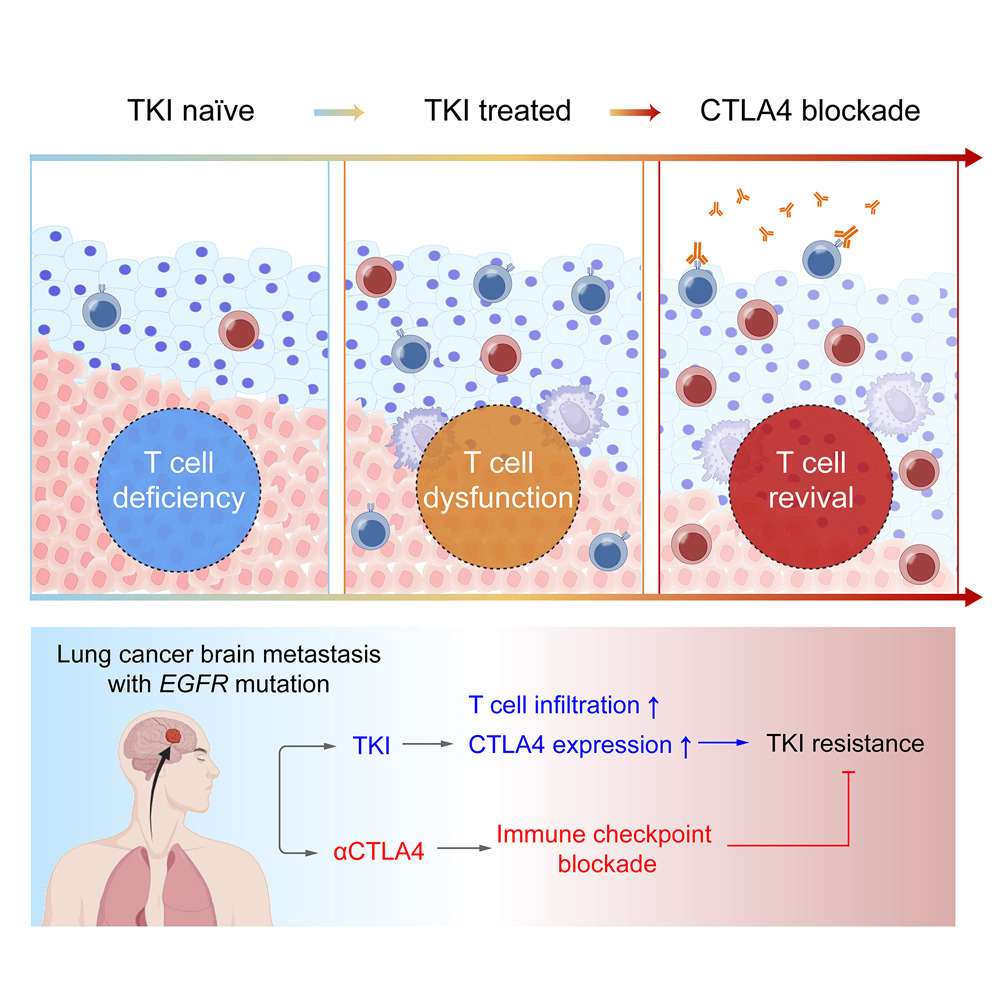
Lung cancer is the leading cause of cancer-related death globally, with approximately 40%-50% of lung cancer patients developing central nervous system (CNS) metastases. The metastatic microenvironment within the CNS, composed of brain-resident and infiltrating immune cells, challenges the traditional view of the CNS as an immune-privileged organ and directly determines the progression and prognosis of lung cancer brain metastasis. Although histopathological diagnosis and imaging techniques have provided important insights into lung cancer brain metastasis, unbiased molecular profiling of the diverse cellular networks across the entire brain remains critical for comprehensively understanding disease progression and biological functions. The development of molecular classification and targeted therapy has brought significant benefits to patients with specific genetic alterations. The advent of tyrosine kinase inhibitors (TKIs), particularly the third-generation EGFR-TKI osimertinib which can penetrate the blood-brain barrier, has revolutionized the treatment landscape for lung cancer brain metastasis, becoming the standard first-line treatment for EGFR-mutant lung cancer brain metastasis. However, acquired TKI resistance remains a major clinical challenge, with most patients experiencing disease progression within one year of treatment. Resistance mechanisms are complex and diverse, with immune evasion being a key driving factor, suggesting that activating anti-tumor immune responses may overcome TKI resistance. Immune checkpoint inhibitors have demonstrated clinical value in various tumors, and some studies have suggested potential for combination strategies between targeted therapy and immunotherapy. However, the efficacy of immune checkpoint blockade in EGFR-mutant lung cancer brain metastasis remains very limited, highlighting the urgent need to investigate the mechanisms of TKI-induced tumor microenvironment reprogramming. To address this, researchers published a study titled "Overcoming tyrosine kinase inhibitor resistance in lung cancer brain metastasis with CTLA4 blockade" in Cancer Cell. This study aimed to systematically analyze the immune microenvironment of lung cancer brain metastasis under different genetic backgrounds and TKI treatment states using single-cell transcriptome sequencing and multiplex immunohistochemistry, confirm CTLA4 as a key target to overcome TKI resistance, and demonstrate in mouse models of EGFR-sensitive and resistant mutant lung cancer brain metastasis that sequential use of EGFR-TKI and CTLA4 blockade is significantly more effective than TKI monotherapy or TKI combined with PD-1 blockade, providing a novel therapeutic strategy for overcoming TKI resistance in lung cancer brain metastasis.
II. Research Methods
This study integrated clinical sample analysis, cellular experiments, animal model validation, and multi-omics technologies. Fresh samples from 31 patients with EGFR-mutant lung cancer brain metastasis (LCBM) and 196 formalin-fixed paraffin-embedded (FFPE) samples were collected. After single-cell suspension preparation, staining, and sorting of fresh samples, single-cell RNA sequencing (scRNA-seq) was performed to conduct high-throughput sequencing of immune cells and tumor cells before and after TKI treatment. Unsupervised clustering analysis was used to identify cell subpopulation composition. Differential gene expression analysis was employed to screen key genes and signaling pathways regulated by TKI and clarify the dynamic changes in the immune microenvironment. For molecular validation, multi-color immunohistochemistry (mIHC) was used to detect protein expression and cellular localization of key molecules including CD3E, CD8A, FOXP3, and CTLA4 in FFPE samples, combined with image analysis software to quantify the proportion of positive cells. Immunofluorescence (IF) was employed to observe changes in HMGB1 nuclear-cytoplasmic distribution. Western blot was used to detect proteins related to NF-κB signaling pathway and CTLA4 expression levels. ELISA was utilized for quantitative detection of HMGB1 in cell culture supernatant. For in vivo experiments, EGFR-sensitive (PC9) and resistant (H1975) cell lines were used to establish lung cancer brain metastasis models in immunodeficient mice. After TKI treatment, CTLA4 blockade was administered sequentially or in combination, and tumor burden and immune cell infiltration were assessed. Flow cytometry was used to detect changes in T cell subpopulations in tumor tissues and peripheral blood.
III. Results and Analysis
1. TKI Reshapes T Cell Differentiation Fate and Induces CTLA4-Mediated Immunosuppression
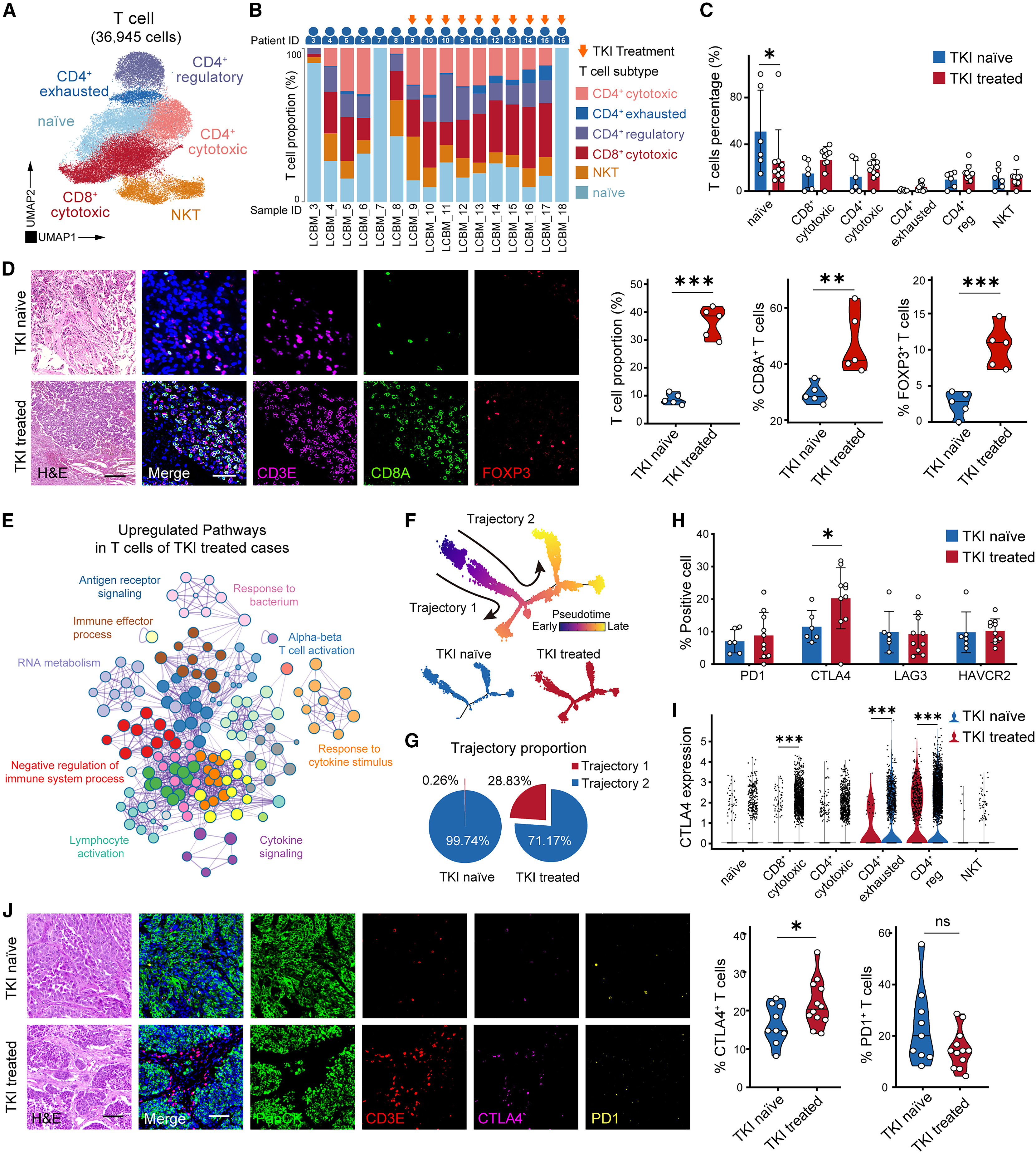
The study first performed cluster analysis of T cells before and after TKI treatment using single-cell sequencing, finding that T cells could be divided into 6 subpopulations. After TKI treatment, the overall infiltration level of T cells was significantly increased, with both CD8+ cytotoxic T cells and CD4+ regulatory T cells showing marked increases in proportion. High infiltration of CD8+ cytotoxic T cells predicted better patient prognosis, while naive T cells and Tregs showed no significant association with prognosis. More importantly, TKI treatment significantly improved prognosis in LCBM patients, but this therapeutic benefit was offset by concurrent immunosuppression—differential gene expression analysis showed that pathways related to lymphocyte activation and cytokine signaling were perturbed in T cells after TKI treatment, while the immune checkpoint molecule CTLA4 was significantly upregulated, and other immune checkpoint molecules such as PD1, TIM3, and LAG3 showed no significant changes.
Further trajectory analysis revealed that T cells have two developmental trajectories (Figure F). In the TKI-treated group, 28.83% of T cells belonged to trajectory 1, while in the untreated group, only 0.26% of T cells belonged to this trajectory. Moreover, CTLA4 expression levels were significantly elevated in the late stage of trajectory 1, suggesting that TKI induces T cell differentiation toward an immunosuppressed phenotype expressing CTLA4. Multi-color immunohistochemistry validation showed that CTLA4 protein levels were significantly elevated after TKI treatment, and were mainly expressed in CD8+ cytotoxic T cells, CD4+ Tregs, and other immune cells. Functional experiments demonstrated that CTLA4+ T cells exhibited impaired effector function, and CTLA4 blockade could restore their anti-tumor activity.
2. Monocyte-Derived Macrophages Mediate T Cell Recruitment After TKI Treatment
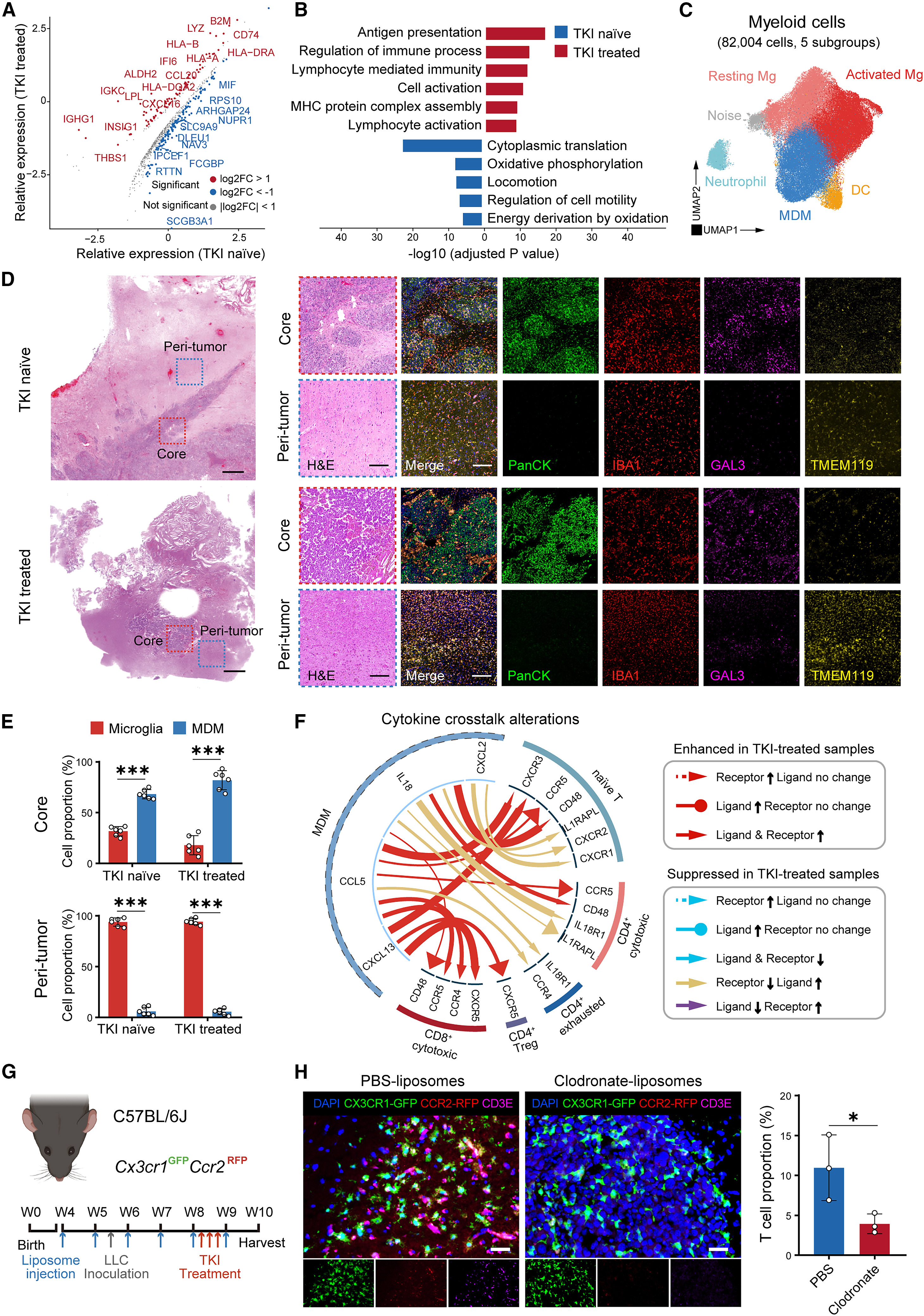
In addition to adaptive immune cells, tumor-associated myeloid cells (TAMs) also play key roles in treatment-induced immune microenvironment reprogramming. The study divided myeloid cells into 5 subpopulations using single-cell sequencing, with monocyte-derived macrophages (MDMs) and microglia being the two most abundant myeloid subpopulations in LCBM, accounting for 32.0% and 56.9%, respectively. Spatial distribution analysis showed that MDMs were more enriched in the tumor core region, while microglia were mainly distributed in the peripheral tumor region, suggesting that MDMs may directly guide T cells into the tumor core.
After TKI treatment, the expression of human leukocyte antigen class II molecules and chemokines in MDMs was significantly elevated. Gene Ontology enrichment analysis showed that antigen presentation and lymphocyte-mediated immune-related pathways were enhanced, indicating that MDMs may participate in TKI-induced T cell recruitment. Cell-cell interaction analysis showed that after TKI treatment, ligand-receptor interactions between MDMs and T cells were significantly enhanced. In mouse models, depletion of peripheral MDMs using clodronate liposomes resulted in a significant reduction in T cell infiltration in the tumor core region. In vitro co-culture experiments further confirmed that conditioned medium from TKI-treated tumor cells could promote T cell migration, with chemokines secreted by MDMs playing a key role. These results indicate that MDMs are the core mediators of TKI-induced T cell infiltration.
3. TKI Induces Immunogenic Cell Death of Tumor Cells and Regulates the Immune Microenvironment Through the HMGB1/TLR/NF-κB Axis
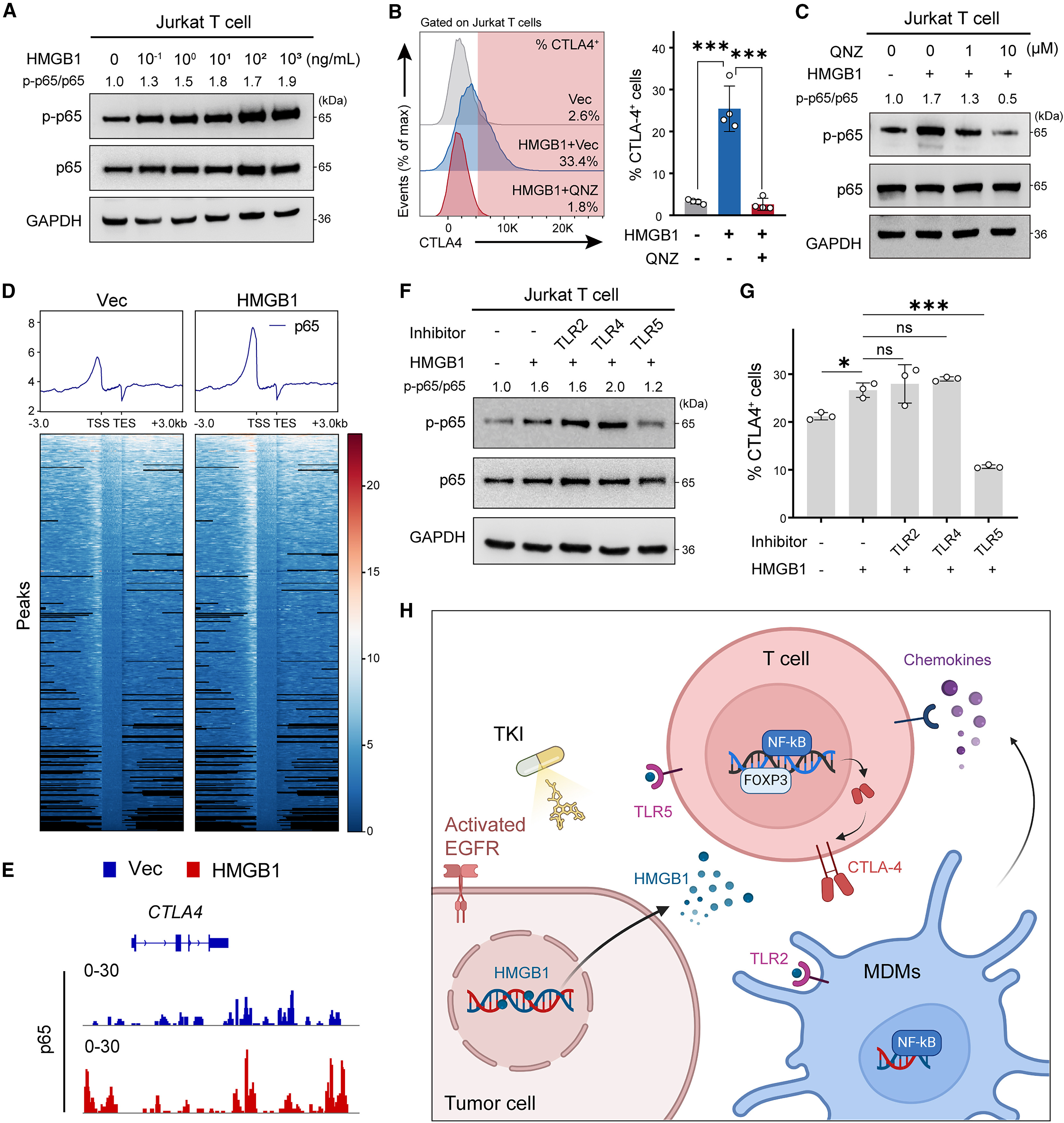
TKI has been demonstrated to induce immunogenic cell death (ICD) in lung cancer cells, and damage-associated molecular patterns (DAMPs) released from ICD are key signals connecting tumor cells with immune cells. The study found that after TKI treatment, tumor cells released high mobility group box 1 (HMGB1) and calreticulin (CALR) among other DAMPs. The nuclear localization ratio of HMGB1 was significantly reduced, with more being released extracellularly in a TKI dose-dependent manner. In vitro experiments showed that HMGB1 could activate the NF-κB signaling pathway in THP1-derived macrophages, while the TLR2 inhibitor (TLR2-IN-C29) could significantly block this activation process and reduce macrophage chemokine release. This suggests that macrophages recognize HMGB1 through TLR2, triggering downstream signaling pathways and recruiting T cells.
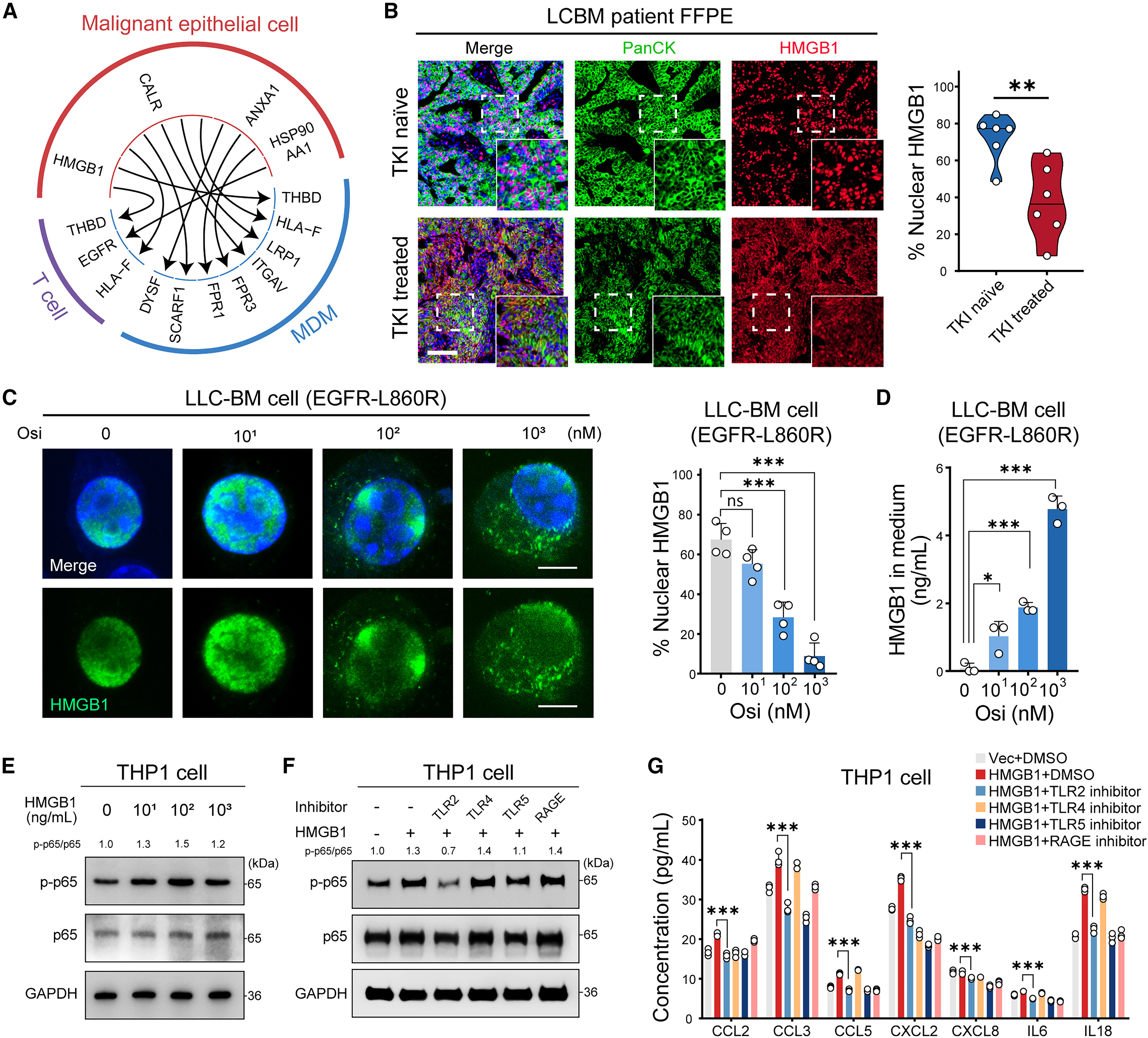
More importantly, HMGB1 can also act on T cells, activating the NF-κB signaling pathway in T cells in a dose-dependent manner, thereby upregulating CTLA4 expression, which can be reversed by the NF-κB inhibitor QNZ. CUT&Tag sequencing showed that HMGB1 promotes NF-κB subunit p65 binding to the CTLA4 promoter region, directly regulating CTLA4 transcription. Further studies found that T cells recognize HMGB1 signals through TLR5, and TLR5 inhibitors can significantly reduce the proportion of CTLA4+ T cells. Thus, the study delineates a complete regulatory pathway: TKI induces tumor cells to release HMGB1, MDMs recognize HMGB1 through TLR2 and release chemokines to recruit T cells, and HMGB1 directly acts on T cells through TLR5 to activate NF-κB signaling and upregulate CTLA4 expression, ultimately leading to T cell dysfunction.
4. CTLA4 Blockade Synergizes with TKI to Reverse Immunosuppression and Improve LCBM Prognosis
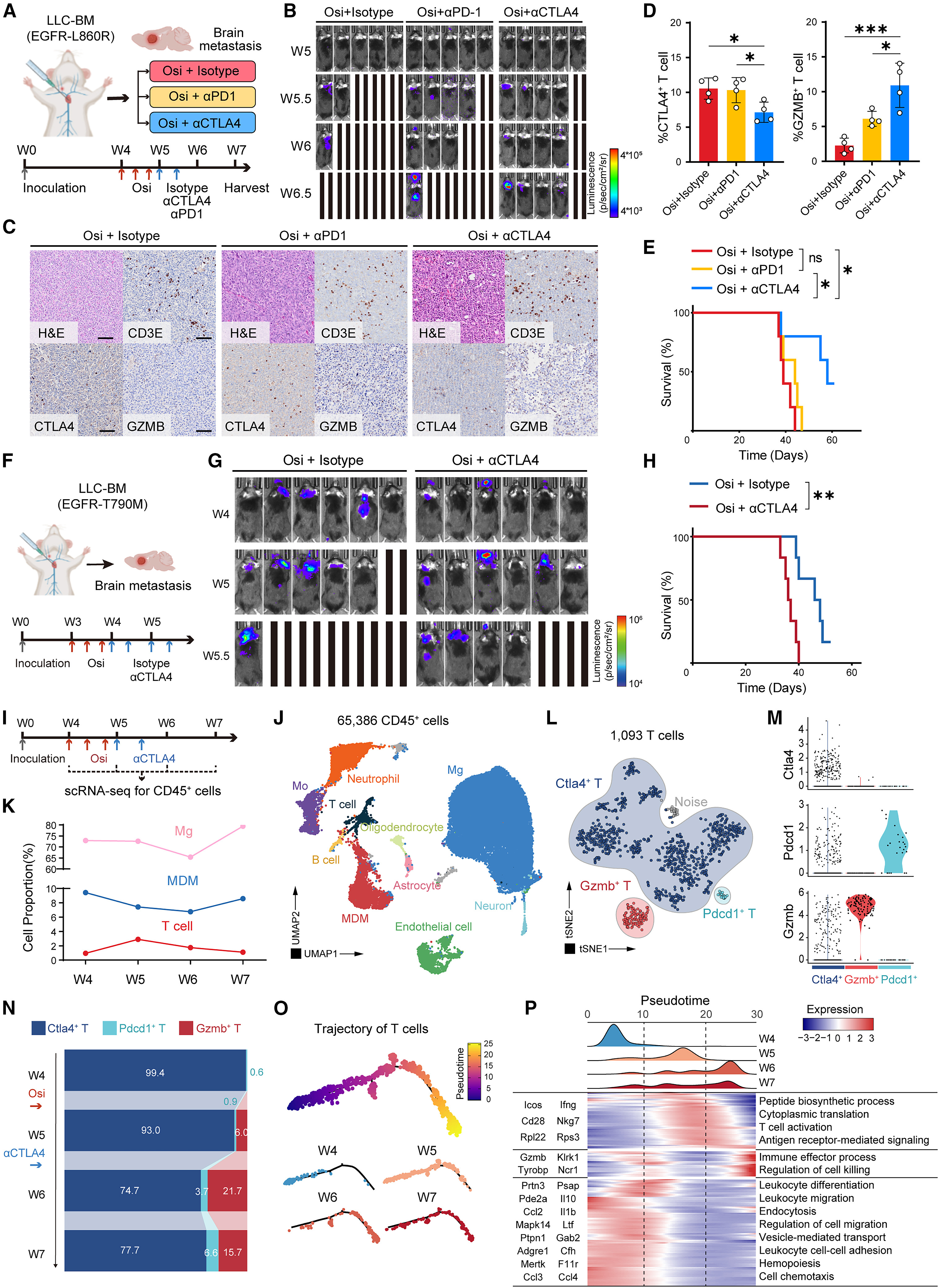
Based on the central role of CTLA4 in TKI-induced immunosuppression, the study further validated the therapeutic effect of TKI combined with CTLA4 blockade. In mouse models of LCBM mimicking human EGFR-L858R mutation, sequential treatment with TKI combined with CTLA4 blockade significantly reduced tumor burden, decreased CTLA4+ T cell proportion, increased GZMB+ effector T cell proportion, and significantly prolonged overall survival compared to TKI monotherapy or TKI combined with PD1 blockade.
In EGFR-T790M mutant LCBM mouse models, TKI combined with CTLA4 blockade also significantly improved mouse survival with good safety, showing no severe hepatotoxicity or pneumonia, and blood biochemical indicators remained within normal ranges. Temporal single-cell sequencing analysis showed that during combined treatment, the immune microenvironment underwent a dynamic transition from T cell dysfunction to T cell recovery: TKI treatment initially activates MDMs to recruit T cells, but simultaneously induces CTLA4 upregulation leading to T cell dysfunction. After adding CTLA4 blockade, the proportion of CTLA4+ T cells decreased, the proportion of GZMB+ effector T cells increased, and the immune effector function of T cells was significantly enhanced, with related gene expression enriched in immune effector processes and cell killing regulatory pathways.
IV. Research Conclusions
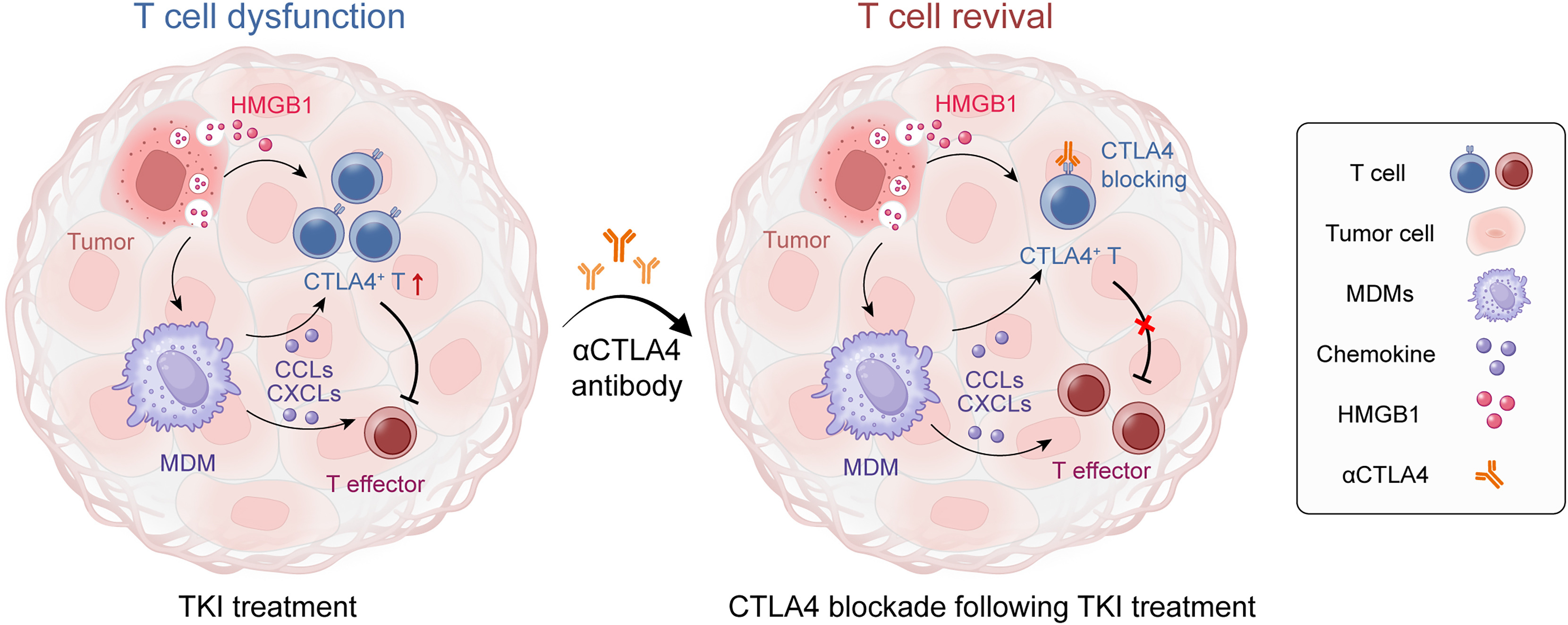
This study for the first time systematically revealed the bidirectional regulatory effect of TKI on the immune microenvironment of EGFR-mutant LCBM: TKI recruits T cells by activating MDMs to address the issue of T cell deficiency, but simultaneously upregulates CTLA4 expression through the HMGB1/TLR/NF-κB axis, inducing T cell dysfunction and ultimately leading to resistance. CTLA4 blockade can effectively reverse this immunosuppression and synergize with TKI to exert anti-tumor effects, showing good therapeutic efficacy in both TKI-sensitive and resistant mutant LCBM. This study challenges the traditional understanding of TKI resistance mechanisms, emphasizes the critical role of immune microenvironment reprogramming in resistance, and provides a new combination strategy for the treatment of EGFR-mutant LCBM, echoing the core theme of the paper's title focusing on overcoming TKI resistance in lung cancer brain metastasis with CTLA4 blockade. In the future, high-quality clinical studies are needed to further validate the clinical efficacy of this combination regimen, while optimizing treatment timing and dosage to bring new therapeutic hope to patients with advanced LCBM.
References
Fu M, Zhao J, Zhang L, Sheng Z, Li X, Qiu F, Feng Y, You M, Xu H, Zhang J, Zeng R, Huang Y, Li C, Chen W, Chen Z, Peng H, Li L, Wu Y, Ye D, Chi Y, Hua W, Mao Y. Overcoming tyrosine kinase inhibitor resistance in lung cancer brain metastasis with CTLA4 blockade. Cancer Cell. 2024 Nov 11;42(11):1882-1897.e7. doi: 10.1016/j.ccell.2024.09.012. Epub 2024 Oct 17. PMID: 39423817.
EnkiLife Products:
Western Blot Complete Experimental Protocol
TSA Multiplex Fluorescence Kit